By
David Bautz, PHD
NASDAQ:CERC
Business Update
Cerecor, Inc. (NASDAQ:CERC) is a clinical stage biopharmaceutical company developing treatments for patients with neurological and psychiatric disorders. The company’s lead compounds include CERC-301, an antagonist of the N-methyl-D-aspartate (NMDA) receptor, CERC-501, a potent and selective antagonist of the kappa opioid receptor (KOR), and CERC-611, a preclinical candidate recently acquired from Eli Lilly for the treatment of epilepsy.
Importantly, Cerecor will announce results from Phase 2 clinical trials of both CERC-301 and CERC-501 in November 2016 and December 2016, respectively. Below we review each compound and the two Phase 2 clinical trials.
CERC-301
CERC-301 is an orally available specific antagonist against the NMDA receptor subunit 2B (NR2B). The compound is being developed as an adjunctive medication in patients with severe major depressive disorder (MDD) who are not responding to their current antidepressant treatment.
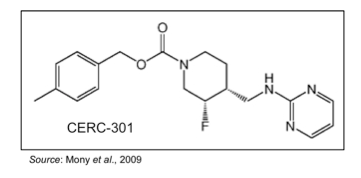
As opposed to currently available MDD treatments that target monoamine neurotransmitters such as serotonin, norepinephrine, and/or dopamine, CERC-301 targets the N-methyl-D-aspartate (NMDA) glutamate receptors. These receptors are composed of various subunits to form three different subtypes: GluN1, GluN2, and GluN3 (or NR1, NR2, and NR3). There are eight variants of the NR1 subunit, four different NR2 subunits (referred to as NR2A through D), and two NR3 subunits (Paoletti et al., 2007). CERC-301 specifically targets the NR2B subunit.
There are a number of recent studies that show the NMDA receptors contribute to the pathophysiology of MDD (Serafini et al., 2013). This may be the result of an imbalance in glutamate signaling that can lead to NMDA agonism, which ultimately leads to enhanced excitatory activity in neural circuits involved in depression. Traditional monoamine antidepressants are known to interfere with glutamate system function by reducing glutamate release and synaptic transmission along with regulating NMDA receptor activity (Musazzi et al., 2013). However, these effects take time, but point to the opportunity to have a rapid effect on depression through NMDA antagonism.
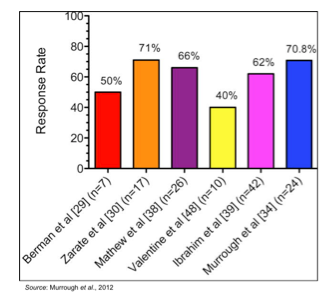
Ketamine, a drug that has been utilized for over 50 years as a dissociative anesthetic, is a known antagonist of the NMDA receptor. It was first studied as a treatment for depression in 2000 in a study involving seven patients with MDD that received a single sub-anesthetic dose (Berman et al., 2000). The results showed significant improvement in depressive symptoms within 72 hours of treatment with ketamine. These results were followed up by additional single-dose studies that showed very similar results (Zarate et al., 2006; Valentine et al., 2011). Multiple-dose studies have also been published showing the efficacy of ketamine in treating MDD (aan het Rot et al., 2010; Murrough et al., 2013). Ketamine also appears to have anti-suicidal properties, as shown in studies involving patients with chronic (DiazGranados et al., 2010) and acute (Larkin et al., 2011) suicide ideation. The effect of ketamine in multiple depression studies is summarized in the following figure, which shows the peak response rates for a series of trials that tested ketamine in patients with MDD (Murrough et al., 2012).
While shown to be quite effective at rapidly reducing depressive symptoms, ketamine has a number of side effects that preclude its widespread use including the fact it is a psychotomimetic that can result in toxicity and a schizophrenic-like syndrome when abused (Behrens et al., 2007). What would be most optimal is a drug that has the rapid anti-depressive action of ketamine but without the toxic side effects.
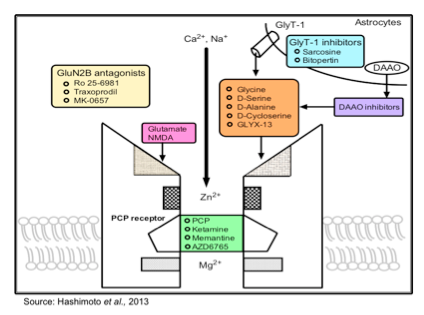
The current understanding about the role of different NMDA receptor subtypes is that compounds that act as subunit-selective modulators are likely to be as efficacious but safer and with less abuse potential than non-selective NMDA receptor antagonists such as ketamine. There are a number of different compounds currently under development that target various NMDA receptor subunits, as shown in the following figure.
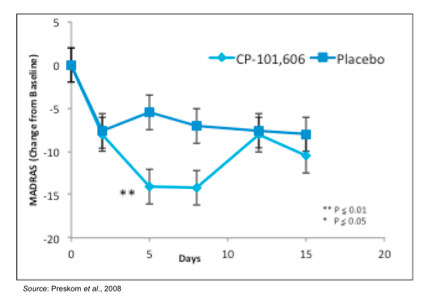
Previous studies with other NR2B-selective antagonists have shown the potential for this class of compounds to be utilized in treating MDD. In 2008, a randomized, double blind clinical trial was performed in patients with MDD who were refractory to standard of care treatment to test CP-101,606, an NR2B-selective antagonist (Preskorn et al., 2008). In this study, patients were first treated in an open-label fashion for six weeks with the SSRI paroxetine followed by a single blind placebo infusion. Those who did not respond to paroxetine (n=30) were then randomized to a single infusion of CP-101,606 or placebo followed by four additional weeks of paroxetine. As the following figure shows, patients treated with CP-101,606 exhibited a greater change in the Montgomery-Asberg Depression Rating Scale (MADRS), which is a scale used by psychiatrists to measure the severity of depressive episodes, than those treated with placebo.
Cerecor initiated a Phase 2 clinical trial of CERC-301 in November 2013 (NCT01941043). The 135-patient, placebo controlled trial consisted of MDD subjects who were resistant to SSRI or SNRI treatment and had recent active suicidal ideation. Patients were treated for 28 days with 8 mg/day of CERC-301 and the primary outcome was the change in the Hamilton Depression Rating Scale (HDRS) on day 7. In March 2015, Cerecor announced that the 8 mg dose used in the study did not meet the primary objective, however the drug was shown to be safe and well-tolerated. In a separate pharmacokinetic/pharmacodynamics study, 48 patients were safely treated with daily doses of CERC-301 of up to 20 mg, thus indicating that higher doses of CERC-301 could be utilized in future clinical trials.
Cerecor is currently conducting the Clin301-203 clinical trial, a Phase 2 randomized, double blind, placebo controlled study evaluating the antidepressant effect of 12 mg and 20 mg doses of CERC-301 in MDD patients currently experiencing a severe depressive episode despite stable ongoing treatment with either an SSRI or SNRI (NCT02459236). In September 2016, Cerecor announced completion of enrollment in the Clin301-203 study with a total of 115 patients. Results from the study will be announced in November 2016.
The primary outcome of the study is the antidepressant effect of both doses of CERC-301 compared to placebo averaged between two and four days’ post-treatment as assessed by the 6-item unidimensional subset of the HDRS known as the Bech-6. This is a subscale of the HDRS that should allow for the detection of acute drug effects along with the duration of the drug effect. Key secondary objectives include evaluating the antidepressant effect of CERC-301 by the full HDRS and the 7-item unidimensional subset known as Santen-7. Additional analyses will include the antidepressant effect at two, four, and seven days after each dose as well as 14 days after the last administration of study drug by the Bech-6, Santen-7, HDRS, Clinically Useful Depression Outcome Scale-Anxiety Self Report (CUDOS-A-SR), and the Snaith-Hamilton Pleasure Scale Self Report (SHAPS-SR). Qualified site raters will administer the clinically-administered tests and the study subjects will administer self-reported scales. There are a total of nine study visits, however four of them will be conducted by phone to alleviate the burden on the subjects. The figure below gives a graphical representation of the study.
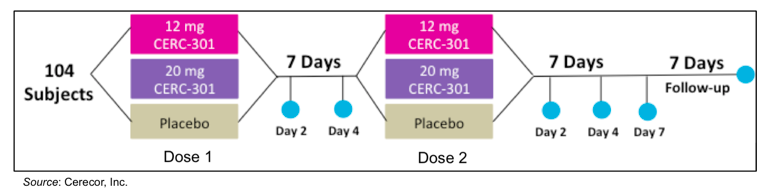
In comparison to the previous Phase 2 trial that Cerecor conducted with CERC-301, there are a number of attributes to the Clin301-203 study that we believe could lead to a greater likelihood of success. Cerecor is testing a higher dose of CERC-301 (12 mg and 20 mg) compared to the first Phase 2 study that only evaluated 8 mg/day. In addition, subjects will take the medication on an empty stomach in order to increase the bioavailability of the drug. Both of these changes will result in maximum drug concentration being two to four times higher in subjects in the Clin301-203 study than in the previous study. Other factors that make the Clin301-203 study more likely to be successful include a more rigorous inclusion/exclusion criteria, recruitment of subjects from psychiatric clinical referrals and depression clinical study databases, and the use of the Bech-6 scale, which is more sensitive to acute changes from fast-acting antidepressants.
The FDA designated CF-301 as a Fast Track product for the treatment of MDD. The Fast Track program was put into place under the FDA Modernization Act of 1997 and is a process designed to facilitate the development, and expedite the review of drugs to treat serious conditions and fill an unmet medical need.
Opportunity in MDD
The treatment of depression represents a significant market opportunity. Approximately 16 million adults in the U.S. will suffer from a major depressive episode during any 12-month period. Of those, approximately 4 million are treated with drug therapy, which is the current target market for CERC-301. Due to the increasing prevalence of generic medications, many ‘Big Pharma’ companies have exited the space or are waiting for smaller biotech companies to produce positive early-stage results before re-entering the market. With an increased understanding of the disease and better treatment target validation, a company such as Cerecor could realize a significant jump in value with positive clinical results.
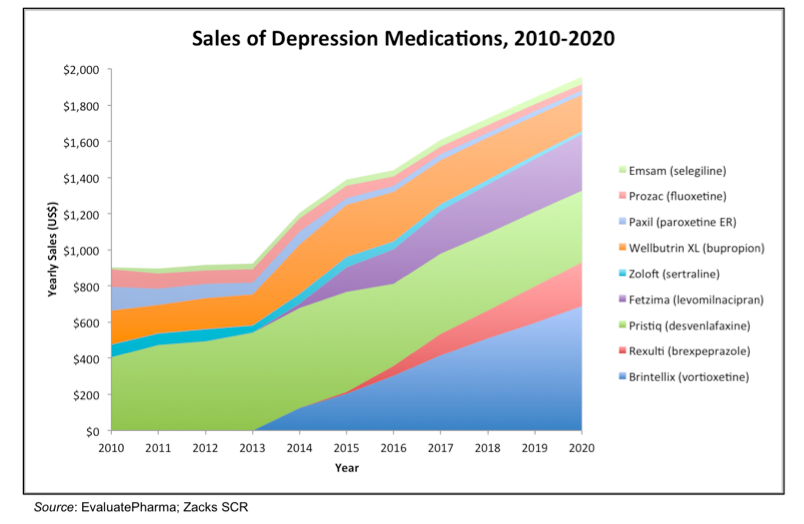
The following chart shows the U.S. sales of different prescription depression medications from 2010-2020. The data after 2015 is from consensus analyst’s forecasts (EvaluatePharma). Growth in the market is going to be supplied by new entrants to the market such as Brintellix, Rexulti, and Fetzima. Even with a number of different treatment options available, there is still plenty of room in the market for an effective treatment with a differentiated mechanism of action.
There are currently five other programs in development by other companies for compounds that are rapid onset antidepressants or anti-suicide treatments:
- Esketamine: This is the S(+) enantiomer of ketamine. It is being developed by Johnson and Johnson as an intranasally administered adjunct treatment for patients with MDD and is currently in a Phase 3 clinical trial.
- Rapastinel: This is an intravenously administered peptide that is a partial agonist of the glycine site of the NMDA receptor being developed as a adjunctive treatment for MDD by Allergan. It recently received Breakthrough Therapy designation from the FDA and Phase 3 clinical trials should be underway soon.
- NRX 1074: This is an orally administered small molecule drug that acts as a partial agonist of the glycine site of the NMDA receptor being developed as a adjunctive treatment for MDD by Allergan. Phase 2 clinical trials are set to get underway in 2016.
- AZD8108: This is an orally administered NMDA receptor antagonist being developed by AstraZeneca. It has completed Phase 1 studies.
- AV-101: This is a prodrug of 7-chloronurenic acid, which is a selective antagonist of the glycine site of the NMDA receptor antagonist and is being developed by VistaGen Therapeutics. The compound is currently in a Phase 2 clinical trial for the treatment of MDD.
- Cyclurad: This is an oral fixed-dose combination of D-cycloserine (an NMDA receptor modulator) and lurasidone (a 5-HT2a receptor antagonist), which is administered following a single dose of ketamine. It is being developed by NeuroRx, Inc. for the treatment of Acute/Elevated Suicide Ideation/Crisis in Bipolar Depression (ASIBD/ESIBD).
CERC-501
Cerecor is developing CERC-501, an orally available, highly specific kappa opioid receptor (KOR) antagonist, as a treatment for substance use disorder, adjunctive treatment of MDD, and potentially for co-occurring disorders. Cerecor acquired the rights to CERC-501, then LY2456302, through an exclusive, worldwide license from Eli Lilly and Company in February 2015.
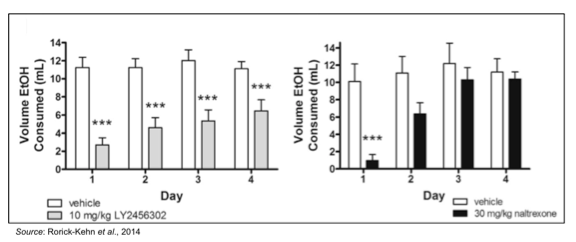
CERC-501 is highly selective for the KOR as it has 21-fold higher affinity for the kappa opioid receptor compared to the mu receptor and 135-fold higher affinity over the delta opioid receptor (Mitch et al., 2011). Preclinical studies showed that CERC-501 dose-dependently produced an antidepressant-like response in the forced swim test and significantly attenuated continuous ethanol self-administration in female rats with a history of high ethanol intake (Rorick-Kehn et al., 2014). The following figure shows that CERC-501 (LY2456302) significantly reduced ethanol consumption on each of the four days of treatment, while naltrexone was only effective on day 1, with tolerance developing by day 2.
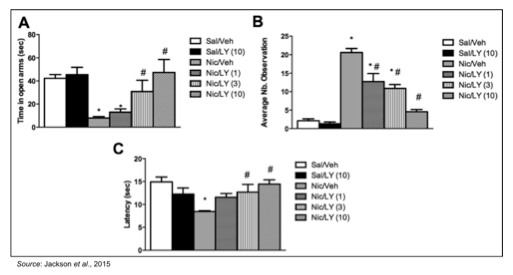
The ability of CERC-501 to treat nicotine withdrawal was studied in an established rodent model where mice were chronically treated with nicotine for two weeks and then studied for withdrawal behaviors 18-24 hours after removal of nicotine with and without CERC-501 (Jackson et al., 2015). The following figure shows that pretreatment with 3 and 10 mg/kg CERC-501 significantly A) decreased the expression of anxiety-related behaviors; B) decreased expression of somatic withdrawal signs; and C) decreased the expression of hyperalgesia. These data support the notion that CERC-501 alleviates physical and affective nicotine withdrawal signs in mice and could be effective in supporting smoking cessation.
The safety, tolerability, and pharmacokinetics of CERC-501 was tested in a Phase 1 clinical trial in healthy subjects (Lowe et al., 2014). Single doses of 2 mg to 60 mg and multiple doses of 2, 10, and 35 mg were administered alone and in combination with ethanol. There were no clinically significant findings and all doses were well tolerated. In addition, there was no evidence for any interaction with ethanol.
Cerecor is currently conducting the Clin501-201 clinical trial, which is a randomized, double blind, placebo controlled trial to evaluate the effects of 15 mg of CERC-501 on tobacco withdrawal and reinstatement and to assess craving, mood, and anxiety during 18 hours of abstinence in 66 heavy cigarette smokers (NCT02641028). Cerecor received a $1 million grant from the National Institute on Drug Abuse to help fund the study. The following graphic gives an overview of the study showing the crossover design, which allows the subjects to be their own control and also significantly increases trial power.
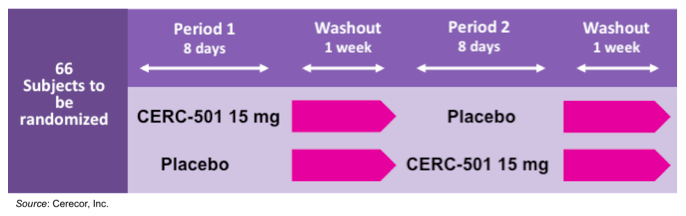
Each period of the trial consists of a seven-day treatment period followed by a single testing day on Day 8. The testing day consists of nicotine deprivation for 18 hours, beginning on the evening of Day 7, and continuing to mid-day of Day 8. This is followed by a smoking lapse test, where subjects are presented with a tray of their preferred brand of cigarettes and an ashtray. The subjects will be told they can smoke as much as they like for 50 minutes (delay period), however for each five-minute block of time a subject delays smoking, they receive a financial reward. Following the smoking of the first cigarette or the end of the 50-minute period, subjects will be provided with eight cigarettes of their preferred brand and allowed to smoke for 60 minutes (self-administration period). The number of cigarettes smoked will be recorded. The primary endpoints of the study are the number of minutes and seconds to the start of tobacco use during the delay period and the number of cigarettes smoked during the self-administration period. In September 2016, the company announced that the trial was fully enrolled with 71 subjects and that data would be reported in the beginning of December 2016.
Opportunity in Treating Nicotine Use
There are approximately 67 million Americans who use tobacco products. Of these, approximately 69%, or 46 million individuals, are interested in quitting. A little more than 50% (24 million) attempted to quit in the past year, and of those only 6% were successful (CDC). Thus, there is a huge unmet medical need for those who would like to quit use of tobacco products but are unable to do it on their own. Currently, there are very few therapeutic options available to help control nicotine addiction. The total market for smoking cessation in 2015 was close to $800 million, which was split approximately 50/50 between over the counter (OTC) medications and Chantix® (varenicline).
Financial Update
On November 8, 2016, Cerecor, Inc. (CERC) announced financial results for the third quarter of 2016 and provided a business update. The company reported approximately $0.3 million in revenue derived from a National Institute on Drug Abuse (NIDA) grant, which provided resources for the ongoing Phase 2 clinical trial of CERC-501. Net loss for the quarter was $6.2 million, or $0.70 per share, and consisted of $4.6 million in R&D expenses and $1.7 million in G&A expenses, compared to $1.3 million in R&D expenses and $0.7 million in G&A expenses for the same time period in 2015. The increase in R&D expenses was mainly due to the $2.0 million upfront payment recorded in connection with the license of CERC-611 in September 2016. Costs associated with CERC-301 and CERC-501 increase by $0.5 million and $0.8 million, respectively, due to increased enrollment in the respective ongoing clinical trials.
At the end of the third quarter 2016, Cerecor had cash and cash equivalents of approximately $8.8 million, which we estimate will be sufficient to fund operations at least through the end of 2016. In September 2016, Cerecor announced it had entered into a $15 million stock purchase agreement with Aspire Capital Fund, LLC. Under terms of the agreement, Aspire made an initial purchase of $1 million on common stock at $4.00 per share and Cerecor will have the right to sell the remaining $14 million of shares to Aspire from time to time over a 30-month period. Cerecor will control the timing and amount of any sales.
As of November 1, 2016, Cerecor had approximately 9.3 million common shares outstanding. In addition, there were approximatley 1.8 million stock options and approximately 7.4 million warrants for a fully diluted share count of 18.6 million.
Conclusion and Valuation
We currently value Cerecor using a probability adjusted discounted cash flow model that takes into account potential future revenues for CERC-301 and CERC-501, with the understanding that this valuation is likely to be greatly affected by the outcome of the two ongoing Phase 2 clinical trials.
For CERC-301, we anticipate that Cerecor will partner the drug and ultimately receive a net 15% royalty on net sales. This takes into account the high single-digit royalty that would be due to Merck upon commercialization of CERC-301. We model for a price of $20 per day in the U.S. and $16 per day in the rest of the world and assume that patients would take the drug for approximately nine months out of the year. Our model calls for a Phase 3 clinical program to get underway in 2018, an NDA filing in 2020, and approval in 2021. We believe approval outside the U.S. would be approximately one year later.
We estimate there are approximately 16 million adults in the U.S. that suffer from MDD each year and approximately 25% of those are treated pharmacologically, which translates to a target population of four million individuals. Outside the U.S., we conservatively estimate another four million individuals are treated with anti-depressants each year. With peak market penetration of 10%, this translates into peak worldwide revenues for CERC-301 of approximately $4 billion. Applying a 10% gross-to-net adjustment, a 96% patient compliance, a 15% net royalty rate, and an 18% discount rate leads to a net present valuation for CERC-301 of $216 million.
For CERC-501, we only include the use of the drug in smoking cessation in our model and have not included any potential revenues from the drug as a treatment for MDD as Cerecor is not conducting trials in that treatment area right now. We anticipate that Cerecor will partner the drug and ultimately receive a net 12% royalty on net sales. This takes into account the tiered royalty rate ranging from mid-single digits to low-double digits that will be due to Eli Lilly upon commercialization of CERC-501. We model for smoking cessation treatment with CERC-501 to cost approximately $180 in the U.S. and $90 outside the U.S. Our model calls for approval of CERC-501 in the U.S. in 2021, and outside the U.S. in 2022.
We estimate there are approximately 55 million smokers in the U.S. and that approximately 45% of those will attempt to quit each year. Outside the U.S., we estimate approximately 80 million smokers with 33% of those attempting to quit each year. We model for peak market penetration of 10% in the U.S. and 5% outside of the U.S., which leads to potential peak worldwide revenues of approximately $500 million. We apply a 12% gross-to-net adjustment, a 96% patient compliance, a net 12% royalty rate, and an 18% discount rate, which leads to a net present valuation for CERC-501 of $21.2 million.
Combining the net present value for CERC-301 and CERC-501 along with the company’s current cash total and expected operating burn of $40 million we arrive at a net present value for the company of $206 million. Dividing this by the company’s fully diluted share count of approximately 18.6 million shares leads to a valuation of approximately $11/share. The stock is currently trading at a significant discount to this valuation, and we believe that positive data from the company’s ongoing Phase 2 clinical trials is likely to cause a significant increase in the share price more in alignment with our valuation.
For a free copy of the full research report, please email scrinvestors@zacks.com with CERC as the subject.
SUBSCRIBE TO ZACKS SMALL CAP RESEARCH to receive our articles and reports emailed directly to you each morning. Please visit our website for additional information on Zacks SCR and to view our disclaimer.