By David Bautz, PhD
NASDAQ:EDSA
READ THE FULL EDSA RESEARCH REPORT
Business Update
Updated Phase 3 ARDS Results Continue to Show Mortality Benefit
On February 24, 2026, Edesa Biotech, Inc. (NASDAQ:EDSA) announced additional Phase 3 results from its paridiprubart (EB05) study in acute respiratory distress syndrome (ARDS) that extend beyond the initial 104-patient cohort that was previously disclosed in October 2025. The updated dataset includes the full 278-patient safety population, which is comprised of both invasive mechanical ventilation (IMV) patients and those who were not on IMV at baseline.
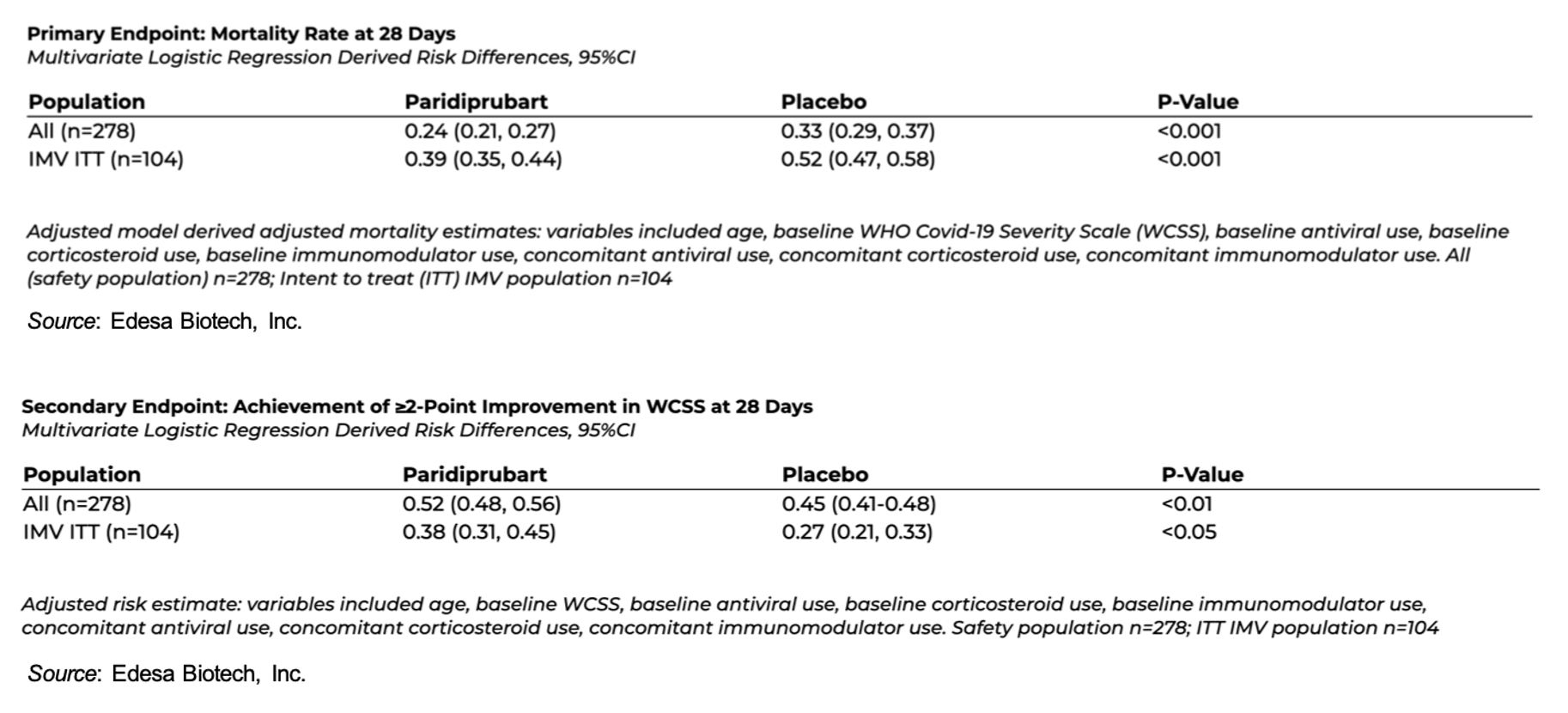
An examination of the full 278-patient population revealed:
- 28-day adjusted mortality was 24% on paridiprubart plus standard-of-care (SOC) compared to 33% on placebo + SOC, which represents a 27% relative reduction in risk of death (P<0.001).
- Patients receiving paridiprubart also demonstrated a higher rate of clinical improvement at Day 28 based on WHO severity scoring.
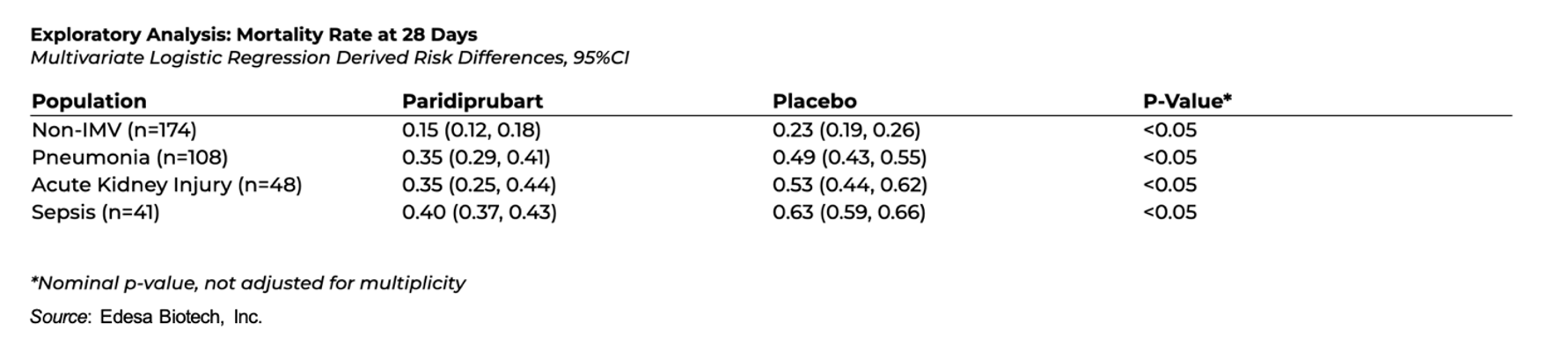
The company also conducted exploratory analyses across clinically relevant subgroups, which suggested patients receiving paridiprubart + SOC consistently had reduced adjusted mortality compared to those receiving placebo + SOC:
- Acute Kidney Injury, n=48: 35% relative reduction (35% vs. 53%; P<0.05)
- Sepsis, n=41: 36% relative reduction (40% vs. 63%; P<0.05)
- Pneumonia, n=108: 30% relative reduction (35% vs. 49%; P<0.05)
Importantly, the safety profile remained consistent compared to prior exposures, with similar rates of adverse events and infections in paridiprubart compared to placebo arms. Over 400 patients have now received paridiprubart.
Given the strength of this data, management has indicated plans to engage with regulatory agencies in both the U.S. and Canada to determine the most appropriate regulatory pathway. Discussions are likely to be focused on whether the robust mortality and clinical improvement signals in the full 278-patient dataset support a registrational submission, the potential for accelerated approval pathway given the high unmet need and the severity of ARDS, and the role of exploratory subgroup data in shaping labeling or accelerated pathways. We anticipate further updates from the company on the regulatory front as the year progresses.
As a reminder, paridiprubart is also being evaluated in an ongoing 200-patient study under funding from BARDA. That study is part of a broader ARDS platform evaluating multiple host-directed therapies and is likely to further inform regulatory decision-making and confirmatory evidence.
Edesa has also been selected to present the data from the Phase 3 study in an oral presentation at the American Thoracic Society (ATS) 2026 International Conference in May 2026, and we anticipate additional findings from the trial at other upcoming medical and scientific conferences.
Phase 2 Vitiligo Trial on Track for Mid-2026 Initiation
Edesa is planning for a Phase 2 study of EB06, its anti-CXCL10 monoclonal antibody, for the treatment of moderate-to-severe non-segmental vitiligo patients. Vitiligo is a disease that causes areas of the skin to lose color, with non-segmental vitiligo being characterized by patches appearing on both sides of the body. It is caused when pigment-producing cells (melanocytes) die or stop producing melanin as a result of an autoimmune disease, genetics, or a triggering event (e.g., stress, sunburn, skin trauma).
Past research showed that the chemokine CXCL10 was elevated in both vitiligo patient skin and serum (El-Domyati et al., 2022). In a mouse model of vitiligo, which includes CXCL10 expression in the skin, neutralization of CXCL10 in mice with established, widespread depigmentation induced reversal of disease as shown by repigmentation (Rashighi et al., 2014). In addition, serum CXCL10 levels are significantly increased in vitiligo patients compared to controls, suggesting that CXCL10 may play a role in the pathogenesis of vitiligo in humans (Gharib et al., 2021). The following slide gives an overview of the mechanism of action of EB06 and data supporting its use in the treatment of vitiligo.
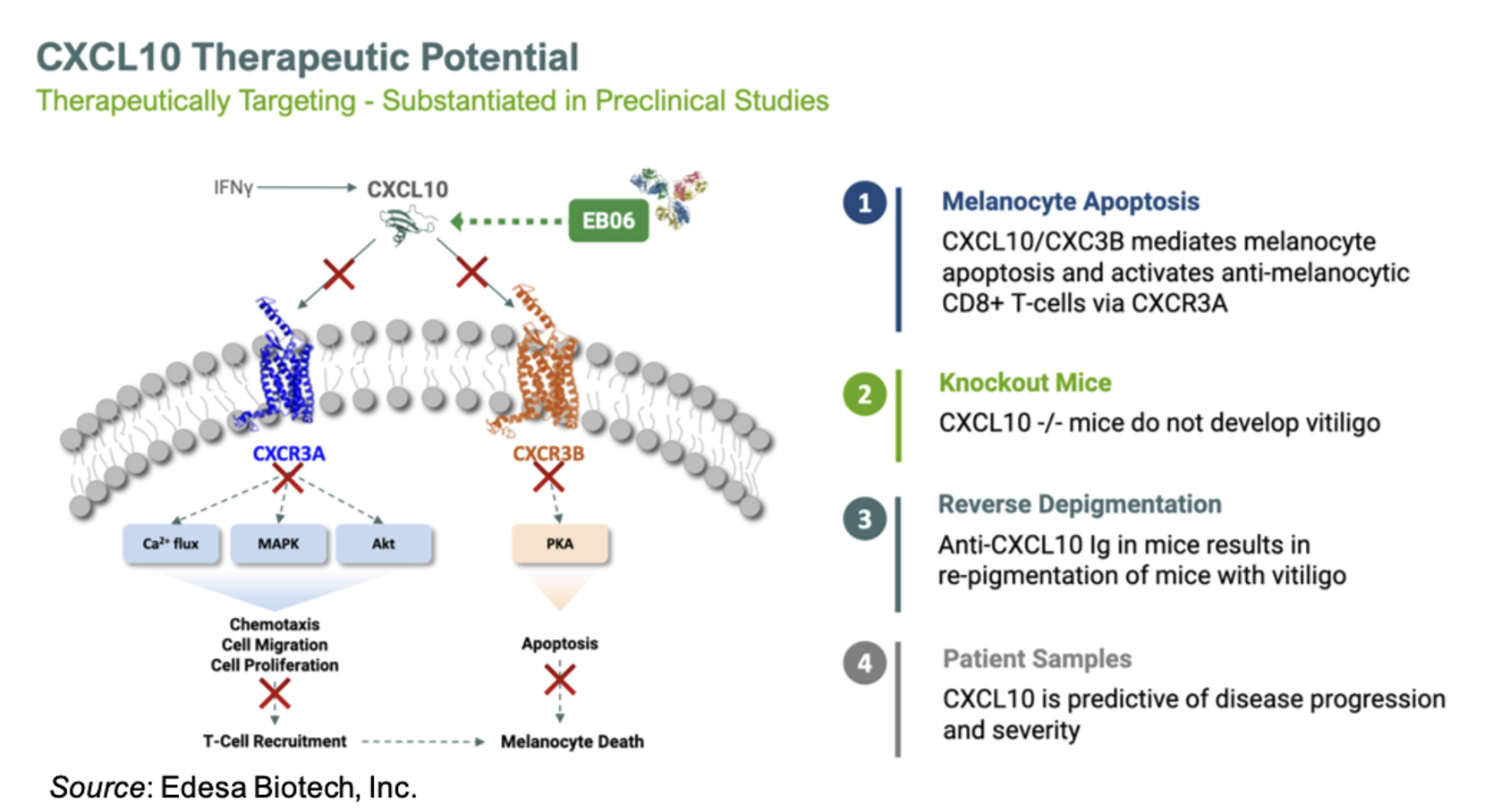
A 2022 publication reported that the estimated prevalence of vitiligo patients in the U.S. is between 1.9 million and 2.8 million (Gandhi et al., 2022). This corresponds to a vitiligo market that is projected to reach approximately $1.1 billion by 2030 (EvaluatePharma). Currently, the only FDA-approved therapy is topical ruxolitinib (Opzelura®), which generated approximately $500 million in revenue in 2024, with approximately $200 million of that coming from sales for vitiligo (EvaluatePharma). Opzelura carries a black-box warning due to the potential for serious infections, major adverse cardiovascular events, and thrombosis (Opzelura prescribing information). Thus, there is clearly an unmet need for additional safe and effective treatment options for vitiligo patients.
Edesa is currently readying an IND submission for EB06 and has already received approval from Health Canada to conduct a Phase 2 trial. In addition, the company has initiated manufacturing activities to supply drug product for the Phase 2 trial. Edesa has also begun outreach to potential investigators. The study as currently planned will enroll approximately 160 patients with severe nonsegmental vitiligo, will evaluate three different doses of EB06 (2.5 mg/kg, 5 mg/kg, 10 mg/kg) administered IV every two weeks for up to 24 weeks followed by a 12-week follow up period, and will have a primary efficacy outcome of the percentage of patients that achieve ≥50% decrease from baseline in facial Vitiligo Area Scoring Index (F-VASI50), a composite measurement of the overall area of facial vitiligo patches and degree of depigmentation within patches. The final trial protocol will be contingent on feedback from the FDA, and we anticipate enrollment initiating in mid-2026, dependent upon completion of manufacturing and regulatory activities.
Financial Update
On February 13, 2026, Edesa announced financial results for the first quarter of fiscal year 2026 that ended December 31, 2025. There were no revenues reported for the first quarter of fiscal year 2026. R&D expenses in the first quarter of fiscal year 2026 were $1.1 million, compared to $1.0 million for the first quarter of fiscal year 2025. The increase was primarily due to higher manufacturing costs and other preparations for a planned Phase 2 clinical study of EB06 in vitiligo patients. G&A expenses totaled $1.2 million for the first quarter of fiscal year 2026 compared to $0.9 million for the first quarter of fiscal year 2025. The increase was primarily due to increased noncash share-based compensation.
As of December 31, 2025, Edesa had approximately $12.1 million in cash and cash equivalents. As of February 12, 2026, Edesa had approximately 8.3 million shares outstanding and, when factoring in stock options, warrants, and the Series B-1 convertible preferred shares, a fully diluted share count of approximately 15.1 million.
Conclusion
The updated Phase 3 results strengthen the case for paridiprubart as a treatment that meaningfully reduces mortality in ARDS across a large safety population, including patients with and without IMV. The drug continues to exhibit an excellent safety and tolerability profile. With no approved pharmacologic treatment for ARDS, these results strongly support discussions with regulatory agencies about the most appropriate pathway, and we look forward to updates from the company as the year progresses. In addition to the strong data for paridiprubart, Edesa remains fully on-track to initiate a Phase 2 study in vitiligo this year, contingent upon regulatory approvals. With no changes to our model, our valuation remains at $19 per share.
SUBSCRIBE TO ZACKS SMALL CAP RESEARCH to receive our articles and reports emailed directly to you each morning. Please visit our website for additional information on Zacks SCR.
DISCLOSURE: Zacks SCR has received compensation from the issuer directly, from an investment manager, or from an investor relations consulting firm, engaged by the issuer, for providing research coverage for a period of no less than one year. Research articles, as seen here, are part of the service Zacks SCR provides and Zacks SCR receives payments totaling a maximum fee of up to $50,000 annually for these services provided to or regarding the issuer. Full Disclaimer HERE.