By John Vandermosten, CFA
NYSE: PLX
READ THE FULL PLX RESEARCH REPORT
2025 Financial and Operational Review
Protalix BioTherapeutics, Inc. (NYSE: PLX) announced 2025 financial and operational results in a March 18th, 2026, press release and Form 10-K filing. The reports were followed by a conference call, which discussed recent achievements, regulatory updates, trial timelines, and financial performance. 2025 revenues fell by 1% as higher Elelyso sales were offset by lower Elfabrio sales, which were impacted by large initial stock-ups in 2024. Protalix and Chiesi requested a re-examination of the Committee for Medicinal Products for Human Use’s (CHMP) negative opinion, which was granted. The agency later issued a positive opinion on four-week dosing. Elfabrio went on to receive European Commission approval in March, which entitles Protalix to a $25 million milestone from Chiesi.
The PRX-115 Phase II trial has launched and is enrolling patients, which is now designated RELEASE. PRX-119 is undergoing pre-investigational new drug (IND) studies this year, and the collaboration continues with Secarna.
Financial results for the year ending December 31st, 2025, compared to the same prior year period:
- Revenues were $52.7 million, down 1% from $53.4 million, attributable to an increase in Elelyso sales to Pfizer offset by a decline in Elfabrio sales to Chiesi. Sales to Pfizer were $18.2 million, up by $5.6 million, and sales to Brazil were a relatively flat $11.1 million. Chiesi sales were $22.5 million, falling by $6.8 million due to the prior year’s inventory build by Chiesi. Protalix also recognized $942,000 in license and R&D services revenue, more than double the prior year amount;
- Cost of revenue was $27.0 million vs. $24.3 million, increasing at a greater rate than revenues;
- Research and development expenses increased to $19.6 million from $13.0 million. Higher salary, subcontractor expenses, materials, and other expenses were up, driving a 51% increase. Increased spending supported preparation activities for the Phase II PRX-115 study, which began in 1Q:26;
- Selling, general, and administrative expenses fell slightly to $11.7 million versus $12.2 million due to a decrease in share-based compensation;
- Net financial income was $108,000 compared to a net financial expense of $237,000. The change in this line item was due to the absence of notes and their associated interest, which were present in 2024;
- Income tax expense of $996,000 vs. $1.2 million;
- Net loss was $6.6 million versus net income of $2.9 million, or -$0.08 per share versus $0.04 per share;
The cash and equivalents balance on December 31st, 2025, totaled $30.3 million versus $34.8 million at the end of 2024. During 2025, Protalix generated $9.3 million from the sale of common stock and exercise of warrants and options. Cash burn was $13.6 million for the year. In March 2026, the EC approved four-week dosing of Elfabrio, qualifying Protalix for a $25 million milestone from Chiesi. We expect the amount will be recognized in the first quarter of 2026, while cash flows will occur in the second quarter.
European Commission Approval of Elfabrio Four Week Dosing
After the back and forth of submissions, responses, and resubmissions, which we have discussed in previous reports, the European Commission (EC) approved the 2.0 mg/kg every four weeks dosing regimen for Elfabrio in Fabry disease. The addressable patient population is adult patients who are stable on enzyme replacement therapy (ERT). Details of the decision were provided in a March 9th, 2026, press release. Along with the approval, Protalix will receive a $25 million milestone payment from Chiesi.
While the four-week dosing regimen has been approved, it will take time to shift patients to it. There are country-specific logistics and regulatory requirements that must be satisfied before patients and providers can make the change.
The Phase III BRIGHT study generated the supportive data to justify extended dosing. Elfabrio offers a prolonged half-life, which enables the change. Adults with Fabry disease already stable on biweekly ERT (agalsidase alfa or beta) for more than three years switched to intravenous pegunigalsidase alfa (Elfabrio) 2.0 mg/kg every 4 weeks for 52 weeks. Kidney function in the stable ERT-experienced group was maintained over a year. There was also an extension to the BRIGHT study, which allowed patients to continue on this regimen. Longer term data from the extension group demonstrated that the change did not increase immunogenicity or create new administration risks.
The three approved ERTs (Fabrazyme, Replagal, and Elfabrio) all require an intravenous (IV) infusion every two weeks, which is a burden that can be reduced by doubling the time between infusions. The change can also reduce provider costs, due to fewer infusions. Other benefits include less venous access trauma, easier scheduling, and a higher quality of life for the Fabry patient.
Background on CHMP Opinion
In December 2024, Protalix’s partner Chiesi submitted a Variation Application to the EMA that requested a change in the dosing regimen for Elfabrio. Based in part on the findings in the BRIGHT study and on new pharmacokinetic data, the sponsors sought a less frequent dosing regimen at a dose of 2 mg/kg body weight administered every four weeks in adult patients with Fabry disease in the European Union. Analysis of the BRIGHT study concluded that treatment with Elfabrio every four weeks could offer a new treatment option for patients with Fabry disease.[1]
On October 17th, 2025, Chiesi and Protalix announced that the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) had issued a negative opinion on the request to approve the dosing regimen of 2.0 mg/kg body weight infused every 4 weeks for Elfabrio.
Two and a half weeks after the negative opinion, Chiesi and Protalix issued a press release stating that they would seek re-examination of the EMA’s negative opinion for Elfabrio regarding the four-week alternative dosing regimen. The process requires that the sponsor submit a written notice to the EMA within 15 days of the CHMP opinion and 60 days later submit the grounds for examination. A different rapporteur and co-rapporteur were appointed to conduct the re-examination. Chiesi and Protalix employed consultants and dedicated internal personnel with EMA and CHMP experience who developed the argument for four-week dosing. Following resubmission, the CHMP gave a positive opinion, and the EC approved the new dosing regimen.
PRX-115 Phase II Trial Enrolling
Protalix announced that the Phase II study for PRX-115 has begun enrolling. The trial, entitled A Study to Investigate the Clinical Effect and the Safety of PRX-115 Infused Intravenously at Different Dosing Regimens, With and Without Methotrexate, Versus Placebo in Adult Gout Patients, is designated RELEASE and is listed on clinicaltrials.gov under NCT07280156. The multicenter, randomized, double-blind, placebo-controlled study will assess the efficacy, safety, and dosing regimen selection of multiple IV infusions of PRX-115 over 24 weeks, with or without methotrexate (MTX), versus the respective placebos in adult patients with gout. One site is up and running in Miami, Florida.
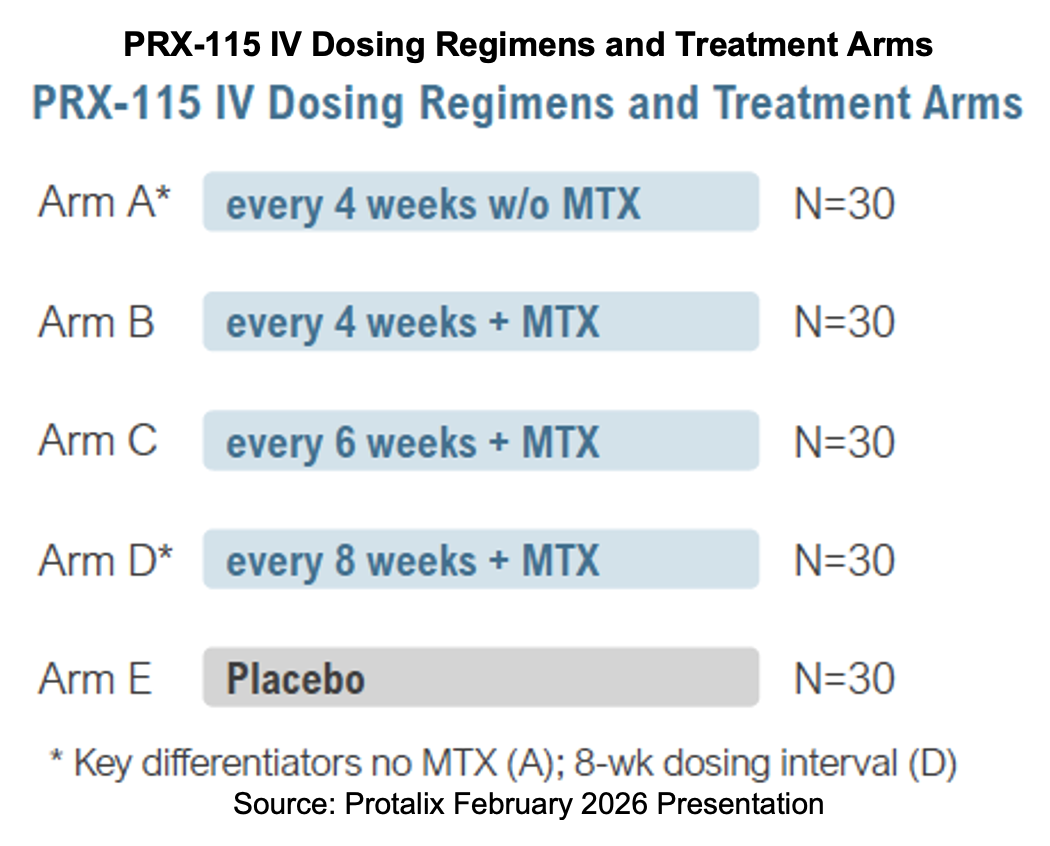
The primary endpoint is the proportion of patients who achieve a reduction in serum uric acid to less than 6.0 mg/dL for at least 80% of the time during month six. Secondary endpoints will measure additional uric acid parameters, safety, and immunogenicity. The study will additionally record tophi, flares, swollen and tender joints, quality of life, and pharmacokinetics.
Participants will receive PRX-115 by intravenous (IV) infusions according to different treatment schedules, with and without MTX. MTX itself does not reduce uric acid or help in managing flares, but is rather used in combination with treatments that may activate antibodies and an immune response. In a study evaluating pegloticase, MTX was used to reduce antidrug antibody development and infusion reactions as well as improve efficacy. The use of MTX with pegloticase improved the response rate for gout patients compared to the use of pegloticase alone.[2]
PRX-115 Background
The PRX-115 Phase I study was completed in 2024, generating data that was presented at conferences, including ACR Convergence. Results from the trial supported advancement to the next stage of development. Planning for the PRX-115 Phase II trial was conducted during 2025, and an investigational new drug (IND) application was filed with the FDA in October. It was cleared in November, and the Phase II started shortly after, enrolling subjects in 1Q:26.
PRX-115 is a plant-cell expressed recombinant PEGylated uricase (urate oxidase) intended to treat uncontrolled gout. Protalix has identified a market of 11.3 million gout patients, with about 300,000 suffering from the uncontrolled form.
SUBSCRIBE TO ZACKS SMALL CAP RESEARCH to receive our articles and reports emailed directly to you each morning. Please visit our website for additional information on Zacks SCR.
DISCLOSURE: Zacks SCR has received compensation from the issuer directly, from an investment manager, or from an investor relations consulting firm, engaged by the issuer, for providing research coverage for a period of no less than one year. Research articles, as seen here, are part of the service Zacks SCR provides and Zacks SCR receives payments totaling a maximum fee of up to $50,000 annually for these services provided to or regarding the issuer. Full Disclaimer HERE.
________________________
[1] Holida, M., et al. A phase III, open-label clinical trial evaluating pegunigalsidase alfa administered every 4 weeks in adults with Fabry disease previously treated with other enzyme replacement therapies, Journal of Inherited Metabolic Disease. October 2024.
[2] Boston, J.K., et al. A Randomized, Placebo‐Controlled Study of Methotrexate to Increase Response Rates in Patients with Uncontrolled Gout Receiving Pegloticase: Primary Efficacy and Safety Findings. Arthritis Rheumatology. December 2022.